CYP450 Interaction Risk Estimator
1. Current Medications
Select drugs known to be strong inhibitors, inducers, or sensitive substrates.
2. Lifestyle & Genetics
Select Factors to Begin
Choose medications and lifestyle factors from the left panel to see your estimated risk level.
Imagine you are taking a statin for your cholesterol and an antibiotic for a sinus infection. On their own, both work fine. Together, they might tear your muscles apart. This isn't magic; it's biology. Specifically, it is a traffic jam in your liver caused by CYP450 enzymes, which are a superfamily of proteins responsible for metabolizing approximately 90% of all clinically used drugs. When medications compete for these enzymes, the results can range from mild side effects to life-threatening toxicity.
We often think of our bodies as static machines, but our metabolism is dynamic. The Cytochrome P450 system acts as the body's primary chemical processing plant. If you understand how this system works, you stop being a passive passenger in your healthcare and start making informed decisions about the prescriptions you take. Let's break down exactly how these interactions happen, why some people are at higher risk, and what you can do to stay safe.
The Liver's Busy Factory: Understanding CYP450 Enzymes
To grasp why drug interactions occur, you first need to know who does the heavy lifting. Your liver is home to thousands of enzymes, but a small group called the Cytochrome P450 (CYP450) family handles the bulk of drug breakdown. These enzymes were named after their unique ability to absorb light at 450 nanometers when bound to carbon monoxide-a quirk discovered in the 1950s that stuck.
Not all CYP450 enzymes are created equal. A few key players dominate the scene:
- CYP3A4: The heavyweight champion. It metabolizes about 50% of all marketed drugs, including many statins, immunosuppressants, and anti-anxiety medications.
- CYP2D6: Handles roughly 25% of drugs, particularly those affecting the heart and brain, like beta-blockers and antidepressants.
- CYP2C9 and CYP2C19: These handle blood thinners like warfarin and clopidogrel, making them critical for cardiovascular health.
- CYP1A2: Processes caffeine, theophylline, and certain antipsychotics.
Think of these enzymes as specialized workers on an assembly line. CYP3A4 is the generalist who picks up any extra work, while CYP2D6 is the specialist focused on specific complex tasks. When too many drugs arrive at once, or when one drug blocks the worker entirely, the assembly line backs up.
How Drugs Compete: Inhibition vs. Induction
Drug interactions via CYP450 generally fall into two camps: inhibition and induction. Both change how much active drug ends up in your bloodstream, but they do so in opposite ways.
Inhibition: Blocking the Exit
Inhibition happens when one drug (the perpetrator) binds to the enzyme and prevents another drug (the victim) from being processed. This is like a truck blocking the only exit ramp off a highway. Traffic piles up behind it.
There are two types of inhibition:
- Reversible (Competitive) Inhibition: The inhibitor temporarily occupies the enzyme's active site. Once the inhibitor leaves, the enzyme can work again. This accounts for 75-80% of significant interactions. For example, if Drug A has a much stronger grip on the enzyme than Drug B, Drug B sits in line longer, leading to higher levels in your blood.
- Irreversible (Mechanism-Based) Inhibition: The inhibitor essentially destroys the enzyme or forms a permanent bond with it. The body must manufacture new enzymes to replace them, which takes 3-7 days. Clarithromycin, a common antibiotic, does this to CYP3A4.
The result? The "victim" drug accumulates. If you are taking simvastatin (a cholesterol drug) and add clarithromycin, your simvastatin levels can skyrocket tenfold within three days. This dramatically increases the risk of rhabdomyolysis, a condition where muscle tissue breaks down and can lead to kidney failure.
Induction: Speeding Up the Line
Induction is the opposite problem. Some drugs signal the liver to produce *more* enzymes. This is like adding more lanes to the highway. While this sounds good, it means drugs are broken down and cleared from your body too quickly.
Rifampin, an antibiotic used for tuberculosis, is a potent inducer of CYP3A4. It can increase enzyme activity by 400-600%. If you are taking birth control pills, blood thinners, or HIV medications alongside rifampin, those drugs may be cleared before they have time to work, leading to treatment failure. Unlike inhibition, induction takes days to weeks to kick in and persists for weeks after you stop the inducing drug.
| Mechanism | Action | Effect on Victim Drug | Onset/Duration | Common Example |
|---|---|---|---|---|
| Inhibition | Blocks enzyme activity | Levels increase (Toxicity risk) | Fast onset; lasts until inhibitor clears | Ketoconazole + Simvastatin |
| Induction | Increases enzyme production | Levels decrease (Treatment failure) | Slow onset (days); persists weeks after stopping | Rifampin + Oral Contraceptives |

The Genetic Lottery: Why You Are Different From Me
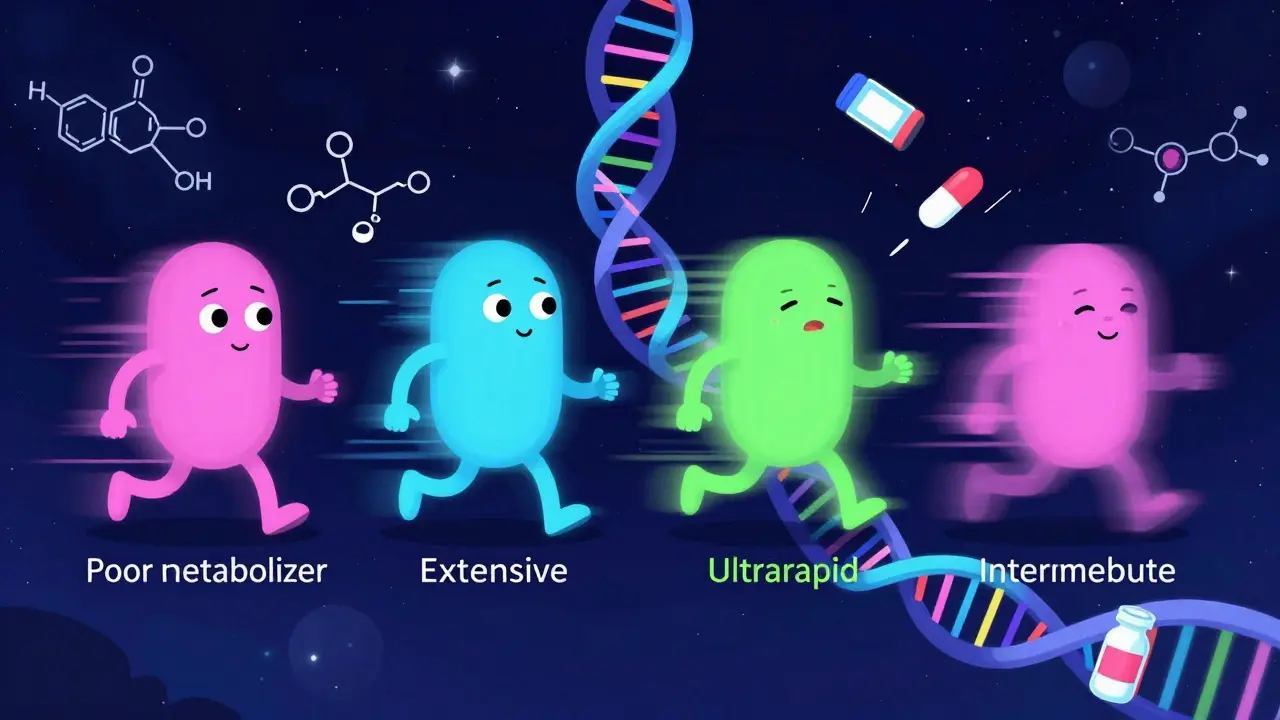
If drug interactions were purely about chemistry, we could predict them perfectly. But humans aren't test tubes. We have genetics. Your DNA determines how many functional copies of each CYP enzyme you produce. This creates four distinct metabolizer phenotypes:
- Poor Metabolizers (PMs): They have little to no functional enzyme activity. Drugs stay in their system longer, increasing toxicity risks. About 5-10% of Caucasians are poor metabolizers for CYP2D6.
- Intermediate Metabolizers (IMs): Reduced enzyme activity. They may need dose adjustments.
- Extensive Metabolizers (EMs): The "normal" majority. They process drugs at expected rates.
- Ultrarapid Metabolizers (UMs): They have multiple copies of the gene, creating extra enzymes. Drugs are cleared too fast, potentially rendering them ineffective.
This genetic variation explains up to 95% of individual differences in drug response. Consider codeine. Codeine is a prodrug-it doesn't work until CYP2D6 converts it into morphine. An ultrarapid metabolizer turns codeine into morphine instantly, risking respiratory depression even at standard doses. A poor metabolizer gets zero pain relief because the conversion never happens. This is why pharmacogenomic testing is becoming standard for drugs like clopidogrel (Plavix), where 30% of Caucasians are intermediate metabolizers with reduced protection against blood clots.
Real-World Risks: Beyond the Pill Bottle
You don't have to be on five different prescriptions to face CYP450 risks. Everyday items can trigger major interactions.
Grapefruit Juice: This is the most famous dietary interaction. Grapefruit contains furanocoumarins that irreversibly inhibit intestinal CYP3A4. Because this enzyme lives in the gut lining, drinking grapefruit juice can reduce the clearance of certain drugs by 30-80%. This affects medications like felodipine (blood pressure), sildenafil (Viagra), and some statins. One glass can have the same effect as doubling your dose.
St. John's Wort: This popular herbal supplement is a powerful inducer of CYP3A4. People taking it for depression often report that their birth control stops working or their transplant rejection meds fail. It induces enzyme activity by 40-60%, mimicking the effects of prescription inducers like rifampin.
Caffeine: If you drink coffee every day, your body induces CYP1A2. If you suddenly stop, or if you take a drug that inhibits CYP1A2 (like fluvoxamine), your caffeine levels can spike, causing jitteriness, insomnia, or even seizures in sensitive individuals. Conversely, smokers induce CYP1A2 significantly, meaning they often need higher doses of drugs like clozapine than non-smokers.
Navigating Polypharmacy Safely
The average Medicare patient takes 5.4 medications daily. With that many drugs, the potential for CYP450 interactions skyrockets. So, how do you protect yourself?
1. Know Your "Index" Drugs: Some drugs have a narrow therapeutic index, meaning the difference between a helpful dose and a toxic one is tiny. Warfarin, digoxin, lithium, and phenytoin fall into this category. If you are taking these, you must be vigilant about interactions.
2. Use Technology: Pharmacists use tools like Lexicomp, which has a 95% sensitivity rate for detecting major interactions. You can access similar checks through patient portals or apps like Epocrates. Always ask your pharmacist to run an interaction check when starting a new medication.
3. Consider Genetic Testing: If you have experienced unexpected side effects or lack of efficacy from standard doses, ask your doctor about pharmacogenomic testing. Panels cost between $250-$500 and can reveal if you are a poor or ultrarapid metabolizer for key enzymes like CYP2D6 and CYP2C19.
4. Disclose Everything: Doctors often focus on prescription drugs. Make sure they know about over-the-counter meds, supplements, and even your diet. St. John's wort, garlic supplements, and green tea can all play roles in enzyme modulation.
The Future of Personalized Dosing
We are moving toward an era where dosing is not one-size-fits-all. The FDA now requires CYP450 interaction data for almost all new drug approvals. Electronic Health Records (EHRs) are integrating real-time alerts that warn prescribers when a dangerous combination is ordered. AI-driven systems are already predicting interactions with high accuracy, helping clinicians navigate complex polypharmacy cases.
However, technology is only as good as the data fed into it. As of 2023, 30% of CYP polymorphisms remain uncharacterized. There is still much to learn about how race, ethnicity, and lifestyle factors influence these enzymes. Until then, awareness is your best defense.
What are the most common drugs that cause CYP450 interactions?
The FDA identifies 27 major drugs known for strong CYP interactions. Common inhibitors include ketoconazole, clarithromycin, and fluoxetine. Common inducers include rifampin, carbamazepine, and St. John's wort. Statins, especially simvastatin and atorvastatin, are frequent victims of these interactions.
Can grapefruit juice interact with all medications?
No, grapefruit juice primarily affects drugs metabolized by CYP3A4, which includes many statins, calcium channel blockers, and anti-anxiety meds. It does not affect drugs processed by other pathways, such as metformin or ibuprofen. Always check your medication label or ask your pharmacist.
How long does it take for a CYP450 interaction to resolve?
For reversible inhibition, effects usually wear off within 1-2 days after stopping the interacting drug. For irreversible inhibition, it can take 3-7 days for the liver to synthesize new enzymes. For induction, effects can persist for 1-3 weeks after discontinuing the inducing agent.
Is pharmacogenomic testing worth it?
It is highly valuable for patients on narrow-therapeutic-index drugs like warfarin, clopidogrel, or certain antidepressants. It can prevent trial-and-error prescribing and reduce adverse events. However, it is less critical for drugs with wide safety margins.
Do age and liver disease affect CYP450 activity?
Yes. Aging naturally reduces liver blood flow and enzyme mass, slowing metabolism. Liver diseases like cirrhosis can severely impair CYP450 function, requiring significant dose reductions for many medications. Children also have developing enzyme systems, making dosing tricky.


